Snakemake is a Pythonic variant of GNU Make. Recently, I learned how to use it to build and launch bioinformatics pipelines on an LSF cluster. However, I had trouble understanding the documentation for Snakemake. I like to learn by trying simple examples, so this post will walk you through a very simple pipeline step by step. If you already know how to use Snakemake, then you might be interested to copy my Snakefiles for RNA-seq data analysis here.
1. Installing Snakemake #
Snakemake is a Python 3 package, so you will need Python 3 installed. It will not work with Python 2.
Go ahead and install snakemake and pyaml with pip3:
pip3 install --user snakemake pyaml
2. Creating a fake workspace with FASTQ files #
In this example, we will process fake paired-end RNA-seq data from FASTQ files. Our “pipeline” consists of two steps:
- Quantify gene expression from the raw RNA-seq reads.
- Collate the gene expression outputs into one master file.
Let’s get started by creating a workspace with our fake data:
cd $HOME
# Create a folder where we will run our commands:
mkdir snakemake-example
cd snakemake-example
# Make a fake genome:
touch genome.fa
# Make some fake data:
mkdir fastq
touch fastq/Sample1.R1.fastq.gz fastq/Sample1.R2.fastq.gz
touch fastq/Sample2.R1.fastq.gz fastq/Sample2.R2.fastq.gz
3. Creating and running a simple Snakefile #
Let’s create a file called Snakefile to complete the first step of our
pipeline. Open your preferred text editor, paste the code below, and save it
into a file called snakemake-example/Snakefile.
SAMPLES = ['Sample1', 'Sample2']
rule all:
input:
expand('{sample}.txt', sample=SAMPLES)
rule quantify_genes:
input:
genome = 'genome.fa',
r1 = 'fastq/{sample}.R1.fastq.gz',
r2 = 'fastq/{sample}.R2.fastq.gz'
output:
'{sample}.txt'
shell:
'echo {input.genome} {input.r1} {input.r2} > {output}'
Understanding the Snakefile #
Let’s walk through the Snakefile line by line.
SAMPLES = ['Sample1', 'Sample2']
We define a list of strings called SAMPLES with our sample names that we’ll
use later in the Snakefile.
rule all:
input:
expand('{sample}.txt', sample=SAMPLES)
The input of rule all represents the final output of your pipeline. In this
case, we’re saying that the final output consists of two files: Sample1.txt
and Sample2.txt. expand() is a special function that is automatically
available to you in any Snakefile. It takes a string like {sample}.txt and
expands it into a list like ['Sample1.txt','Sample2.txt'].
rule quantify_genes:
input:
genome = 'genome.fa',
r1 = 'fastq/{sample}.R1.fastq.gz',
r2 = 'fastq/{sample}.R2.fastq.gz'
output:
'{sample}.txt'
shell:
'echo {input.genome} {input.r1} {input.r2} > {output}'
Because we specified Sample1.txt and Sample2.txt as the final output
files, we need a rule for how to create these files. Instead of writing two
rules (one rule for Sample1 and a second rule for Sample2) we write just one
rule with the special string {sample}.txt as the output.
When Snakemake reads {sample}.txt, it knows to replace it with each of the
values inside SAMPLES to create Sample1.txt and Sample2.txt. Next, it
will extract Sample1 from the string Sample1.txt and put it into the input
files. So, fastq/{sample}.R1.fastq.gz becomes fastq/Sample1.R1.fastq.gz
and fastq/{sample}.R2.fastq.gz becomes fastq/Sample1.R2.fastq.gz.
In our fake pipeline, we won’t actually quantify gene expression. Instead, we’ll just echo the names of the input files into an output file.
Running the pipeline #
We can run the pipeline by invoking snakemake. It knows to look for a file
called Snakefile. Otherwise, you can specify a file to use with the
--snakefile option.
snakemake
Provided cores: 1
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 all
2 quantify_genes
3
rule quantify_genes:
input: fastq/Sample2.R1.fastq.gz, genome.fa, fastq/Sample2.R2.fastq.gz
output: Sample2.txt
1 of 3 steps (33%) done
rule quantify_genes:
input: fastq/Sample1.R1.fastq.gz, genome.fa, fastq/Sample1.R2.fastq.gz
output: Sample1.txt
2 of 3 steps (67%) done
localrule all:
input: Sample1.txt, Sample2.txt
3 of 3 steps (100%) done
Here are the output files that were created:
head Sample?.txt
==> Sample1.txt <==
genome.fa ./fastq/Sample1.R1.fastq.gz ./fastq/Sample1.R2.fastq.gz
==> Sample2.txt <==
genome.fa ./fastq/Sample2.R1.fastq.gz ./fastq/Sample2.R2.fastq.gz
We can create a graphical representation of the pipeline like so:
snakemake --forceall --dag | dot -Tpng > dag1.png
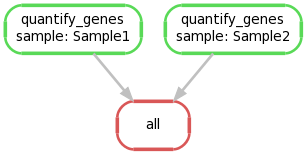
4. Extending the Snakefile to collate output files #
Let’s extend our Snakefile to have one more rule. We’ll collate the two output files into one master file that represents all samples.
Here’s the new modified Snakefile. Notice that the final output for our
pipeline (specified in the rule all section) is now called test.txt. Also
notice that we have a recipe for creating the test.txt file in rule collate_outputs.
rule all:
input:
'test.txt'
rule quantify_genes:
input:
genome = 'genome.fa',
r1 = 'fastq/{sample}.R1.fastq.gz',
r2 = 'fastq/{sample}.R2.fastq.gz'
output:
'{sample}.txt'
shell:
'echo {input.genome} {input.r1} {input.r2} > {output}'
rule collate_outputs:
input:
expand('{sample}.txt', sample=SAMPLES)
output:
'test.txt'
run:
with open(output[0], 'w') as out:
for i in input:
sample = i.split('.')[0]
for line in open(i):
out.write(sample + ' ' + line)
Understanding the Snakefile #
rule all:
input:
'test.txt'
We no longer need the {sample}.txt files in rule all, because the final
output test.txt depends on those intermediate files in rule collate_outputs. Snakemake will figure out that it needs to create the
{sample}.txt files before it creates the final test.txt file.
rule collate_outputs:
input:
expand('{sample}.txt', sample=SAMPLES)
output:
'test.txt'
run:
with open(output[0], 'w') as out:
for i in input:
sample = i.split('.')[0]
for line in open(i):
out.write(sample + ' ' + line)
This rule uses Python code to collate the output files from quantify_genes
into one master file. We read the Sample1.txt and Sample2.txt files line
by line and append the sample name and a single space before the original
content. I use this design pattern for real outputs from real bioinformatics
pipelines, and it also works for our fake pipeline here.
Running the pipeline #
snakemake
Provided cores: 1
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 all
1 collate_outputs
2
rule collate_outputs:
input: Sample1.txt, Sample2.txt
output: test.txt
1 of 2 steps (50%) done
localrule all:
input: test.txt
2 of 2 steps (100%) done
This is the output:
cat test.txt
Sample1 genome.fa ./fastq/Sample1.R1.fastq.gz ./fastq/Sample1.R2.fastq.gz
Sample2 genome.fa ./fastq/Sample2.R1.fastq.gz ./fastq/Sample2.R2.fastq.gz
Again, we can create a graphical representation of the pipeline like so:
snakemake --forceall --dag | dot -Tpng > dag2.png
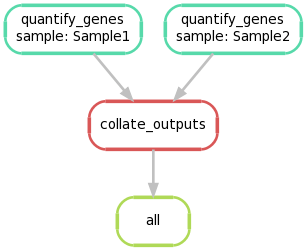
5. Extending the Snakefile to use regular expression glob strings #
Previously, we hard-coded the sample names like this:
SAMPLES = ['Sample1', 'Sample2']
For real work, we might want to make our Snakefile more flexible by using regular expressions to capture sample names from file names.
Below, we have extended the Snakefile to use regular expression glob strings:
from os.path import join
# Globals ---------------------------------------------------------------------
# Full path to a FASTA file.
GENOME = 'genome.fa'
# Full path to a folder that holds all of your FASTQ files.
FASTQ_DIR = './fastq/'
# A Snakemake regular expression matching the forward mate FASTQ files.
SAMPLES, = glob_wildcards(join(FASTQ_DIR, '{sample,Samp[^/]+}.R1.fastq.gz'))
# Patterns for the 1st mate and the 2nd mate using the 'sample' wildcard.
PATTERN_R1 = '{sample}.R1.fastq.gz'
PATTERN_R2 = '{sample}.R2.fastq.gz'
# Rules -----------------------------------------------------------------------
rule all:
input:
'test.txt'
rule quantify_genes:
input:
genome = GENOME,
r1 = join(FASTQ_DIR, PATTERN_R1),
r2 = join(FASTQ_DIR, PATTERN_R2)
output:
'{sample}.txt'
shell:
'echo {input.genome} {input.r1} {input.r2} > {output}'
rule collate_outputs:
input:
expand('{sample}.txt', sample=SAMPLES)
output:
'test.txt'
run:
with open(output[0], 'w') as out:
for i in input:
sample = i.split('.')[0]
for line in open(i):
out.write(sample + ' ' + line)
Understanding the Snakefile #
from os.path import join
You can include any Python code inside your Snakefile, including import
statements to use functions from other packages.
# Full path to a folder that holds all of your FASTQ files.
FASTQ_DIR = './fastq/'
# A Snakemake regular expression matching the forward mate FASTQ files.
SAMPLES, = glob_wildcards(join(FASTQ_DIR, '{sample,Samp[^/]+}.R1.fastq.gz'))
# Patterns for the 1st mate and the 2nd mate using the 'sample' wildcard.
PATTERN_R1 = '{sample}.R1.fastq.gz'
PATTERN_R2 = '{sample}.R2.fastq.gz'
You might notice that SAMPLES, has a trailing comma. It turns out that you
must include this trailing comma, or else the code won’t work correctly.
The glob_wildcards() function is a function similar to glob.glob() (see
here), but it takes a special syntax. It will match all the .fastq.gz
files inside ./fastq/ that match the regular expression
Samp[^/]+.R1.fastq.gz. Also, the part in curly brackets {} will be saved,
so the variable SAMPLES is a list of strings ['Sample1','Sample2'].
r1 = join(FASTQ_DIR, PATTERN_R1),
r2 = join(FASTQ_DIR, PATTERN_R2)
If we evaluate the code join(FASTQ_DIR, PATTERN_R1), we get
./fastq/{sample}.R1.fastq.gz. By using variables instead of hard-coding the
path, we gain some flexibility to customize this Snakefile for new datasets.
6. Launching jobs on an LSF cluster. #
So far, we’ve been running Snakemake without any job scheduler. To launch jobs
in a queue on LSF, you can invoke snakemake like this:
snakemake --jobs 999 --cluster 'bsub -q normal -R "rusage[mem=4000]"'
This will launch up to 999 jobs on the normal queue and request 4 GB of
memory for each job. When you have a long chain of dependencies with multiple
jobs, Snakemake will wait for the dependencies to complete before launching
the next job, as appropriate.
