Count the number of coding base pairs in each Gencode gene
2013-12-23

We can use Python to count the coding base pairs in each Gencode gene. Here, we report the base pair count by gene rather than by transcript. When we encounter different transcripts for the same gene with overlapping exons, we only count those base pairs once rather than multiple times.
Download the gencode coordinates:
ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_19/gencode.v19.annotation.gtf.gz
chr1 HAVANA gene 11869 14412 . + . gene_id "ENSG00000223972.4";
chr1 HAVANA transcript 11869 14409 . + . gene_id "ENSG00000223972.4";
chr1 HAVANA exon 11869 12227 . + . gene_id "ENSG00000223972.4";
chr1 HAVANA exon 12613 12721 . + . gene_id "ENSG00000223972.4";
chr1 HAVANA exon 13221 14409 . + . gene_id "ENSG00000223972.4";
Also, download the NCBI mappings from Entrez GeneID to Ensembl identifiers:
ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2ensembl.gz
9606 1 ENSG00000121410 NM_130786.3 ENST00000263100 NP_570602.2 ENSP00000263100
9606 2 ENSG00000175899 NM_000014.4 ENST00000318602 NP_000005.2 ENSP00000323929
9606 3 ENSG00000256069 NR_040112.1 ENST00000543404 - -
9606 9 ENSG00000171428 NM_000662.5 ENST00000307719 NP_000653.3 ENSP00000307218
9606 9 ENSG00000171428 XM_005273679.1 ENST00000517492 XP_005273736.1 ENSP00000429407
Run the script below to get the following output:
python coding_lengths.py \
-g gencode.v19.annotation.gtf.gz -n gene2ensembl.gz -o output.gz
Ensembl_gene_identifier GeneID length
ENSG00000000005 64102 1339
ENSG00000000419 8813 1185
ENSG00000000457 57147 3755
ENSG00000000938 2268 3167
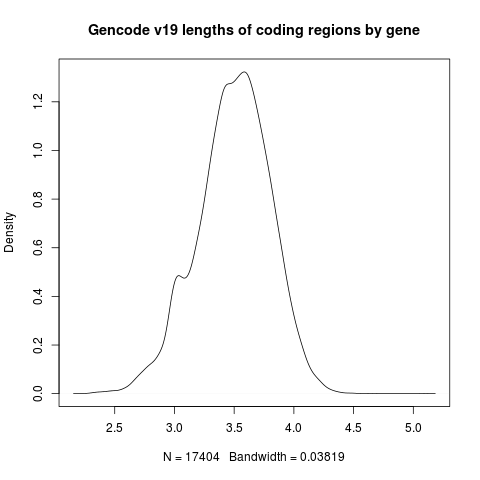
We can plot the output with R like so:
d = read.delim("output.gz")
png("coding_lengths.png")
plot(density(log10(d$length)),
main="Gencode v19 lengths of coding regions by gene")
dev.off()
Source code#
Download coding_lengths.py
Please note that this code requires a class called GTF, defined in GTF.py